El síndrome de Schwartz-Jampel (SJS) es un trastorno hereditario raro que causa anormalidades de los músculos esqueléticos.
Algunas de las anomalías causadas por el trastorno incluyen las siguientes: my Miopatía miotónica (debilidad y rigidez muscular) dys Displasia ósea (cuando los huesos no se desarrollan normalmente)
- Contracturas articulares (las articulaciones se fijan en su lugar, restringiendo el movimiento)
- Enanismo ( estatura baja)
- SJS se considera principalmente una condición autosómica recesiva, lo que significa que un individuo necesita heredar dos genes defectuosos, uno de cada padre, para desarrollar el síndrome.
- Tipos de síndrome de Schwartz-Jampel
Existen varios subtipos del síndrome de Schwartz-Jampel. Tipo I, considerado el tipo clásico, tiene dos subtipos que se han rastreado a un gen defectuoso en el cromosoma 1:
Tipo IA se vuelve aparente más tarde en la infancia y es menos grave.
El tipo IB es aparente inmediatamente al nacer y tiene síntomas más severos.
- El síndrome de Schwartz-Jampel Tipo II es aparente inmediatamente al nacer. Tiene síntomas algo diferentes que IA o IB y no está asociado con ningún defecto genético en el cromosoma uno.
- Por lo tanto, muchos expertos creen que el Tipo II es en realidad la misma enfermedad que el síndrome de Stuve-Wiedermann, un trastorno esquelético raro y grave con una alta tasa de muerte en los primeros meses de vida, principalmente debido a problemas respiratorios.
Por lo tanto, Tipo I será el foco de este artículo.
Síntomas de SJS
El síntoma principal del síndrome de Schwartz-Jampel es la rigidez muscular. Esta rigidez es similar a la del Síndrome de la persona rígida o Síndrome de Isaacs, pero la rigidez del síndrome de Schwartz-Jampel no se alivia con la medicación o el sueño. Los síntomas adicionales de SJS pueden incluir:
Una estatura baja
Rasgos de rasgos faciales, esquinas estrechas de los ojos y una mandíbula inferior pequeña de Deformidades en las articulaciones, como cuello corto, curvatura hacia afuera de la columna vertebral (cifosis) o pecho sobresaliente (pectus carinatum) , también llamado cofre de paloma) Anormalidades del crecimiento de hueso y cartílago (esto se denomina condrodistrofia) Muchas personas con SJS también tienen varias anomalías oculares (ocular), lo que produce diversos grados de discapacidad visual Es importante comprender que cada El caso SJS es único y varía en el rango y la gravedad de los síntomas asociados, según el tipo de trastorno.Cómo se diagnostica la condición
- SJS generalmente se detecta durante los primeros años de vida, con mayor frecuencia al nacer. Los padres pueden notar los músculos rígidos de un bebé durante los cambios de pañal, por ejemplo. Esta rigidez más las características faciales comunes al síndrome a menudo apuntan al diagnóstico.
- Se realizarán más estudios como rayos X, biopsia muscular, análisis de sangre con enzimas musculares y pruebas de conducción muscular y nerviosa en el niño para confirmar anomalías consistentes con SJS. Las pruebas genéticas para el gen defectuoso en el cromosoma 1 (el gen HSPG2) también pueden confirmar el diagnóstico.
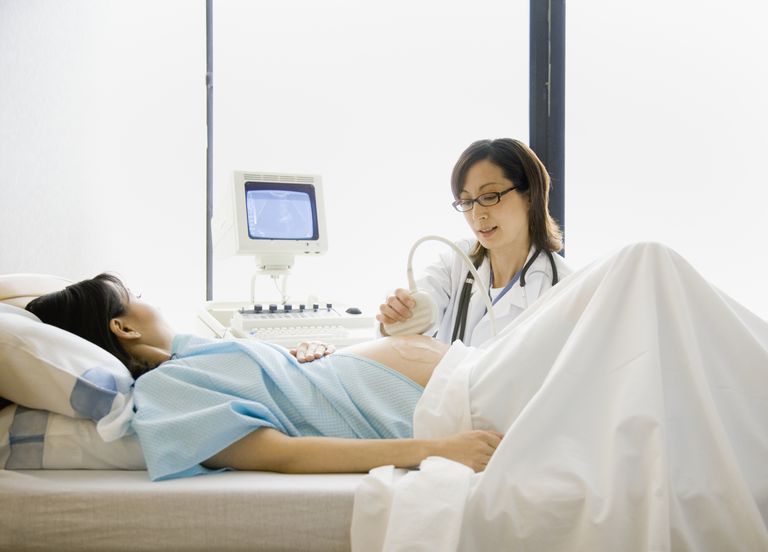
- En algunos casos raros, el diagnóstico prenatal (antes del nacimiento) de SJS puede ser posible mediante el uso de un ultrasonido para ver si el feto tiene características que apuntan a SJS u otras anormalidades del desarrollo.
- Causas del síndrome de Schwartz-Jampel
- Dado que SJS se hereda principalmente de forma autosómica recesiva, si un individuo nace con el síndrome, ambos padres son portadores del gen defectuoso. Cada futuro hijo que tengan estos padres tendrá una probabilidad de 1 en 4 de nacer con el síndrome. En raras ocasiones, se cree que SJS se hereda con un patrón autosómico dominante. En estos casos, solo se necesita heredar un gen defectuoso para que la enfermedad se manifieste.
En general, SJS es un trastorno raro con solo 129 casos registrados, según un informe en
Advanced Biomedical Research
. SJS no acorta la esperanza de vida, y los hombres y las mujeres se ven afectados por igual por la enfermedad.
SJS tipo II (denominado síndrome de Stuve-Wiedermann) parece ser más común en individuos de ascendencia de los Emiratos Árabes Unidos.
Tratamiento del síndrome de Schwartz-Jampel
No hay cura para el síndrome de Schwartz-Jampel, por lo que el tratamiento se enfoca en reducir los síntomas del trastorno. Los medicamentos que son útiles en otros trastornos musculares, como el medicamento anticonvulsivo Tegretol (carbamazepina) y la medicación antiarrítmica mexiletina pueden ser útiles.
Sin embargo, la rigidez muscular en el síndrome de Schwartz-Jampel puede empeorar lentamente con el tiempo, por lo que puede ser preferible utilizar otros medicamentos además del medicamento. Estos incluyen masajes musculares, calentamiento, estiramiento y calentamiento antes del ejercicio.
La cirugía para tratar o corregir anormalidades musculoesqueléticas, como contracturas articulares, cifoescoliosis (donde la columna vertebral se curva de forma anormal) y displasia de cadera puede ser una opción para algunos pacientes con SJS. Para algunos, la cirugía combinada con terapia física puede ayudar a mejorar la capacidad de caminar y realizar otros movimientos de forma independiente.Para problemas visuales y oculares, cirugía, lentes correctivos, lentes de contacto, Botox (para espasmos del párpado) u otros métodos de apoyo pueden ayudar a mejorar la visión.