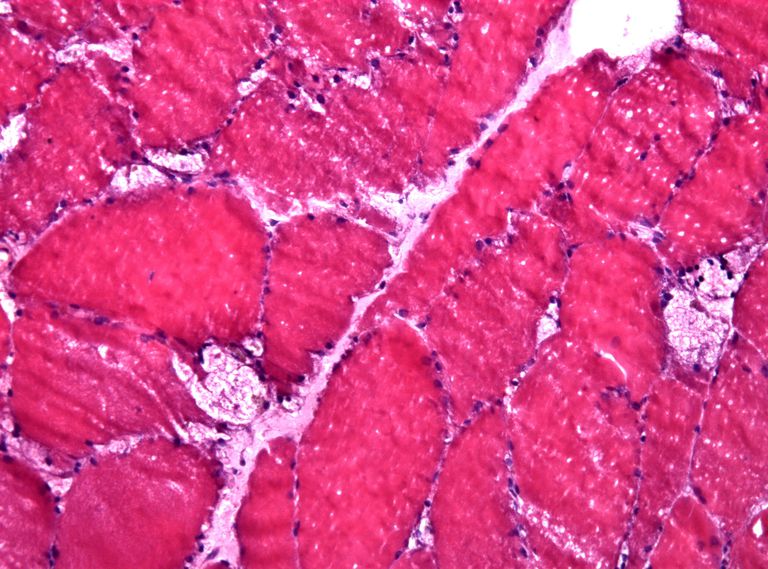
Enfermedad de Pompe o Enfermedad de Almacenamiento de Glucógeno II (GSD-II)
La enfermedad de Pompe, también conocida como enfermedad de almacenamiento de glucógeno tipo II (GSD-II) o deficiencia de maltasa ácida, es uno de los 49 trastornos de almacenamiento lisosómico conocidos.
El nombre de enfermedad de Pompe proviene del patólogo holandés J.C. Pompe, quien describió por primera vez a un bebé con la enfermedad en 1932. La enfermedad de Pompe afecta a entre 5,000 y 10,000 personas en todo el mundo. En los Estados Unidos, se estima que afecta a 1 por 40,000 personas.
La enfermedad de Pompe es causada por una deficiencia o falta completa de una enzima llamada alfa-glucosidasa ácida. Si esta enzima no funciona correctamente, el glucógeno, un azúcar complejo, se acumula en las células del cuerpo y causa daños a órganos y tejidos. Esta acumulación afecta principalmente a los músculos del cuerpo, lo que lleva a la debilidad muscular generalizada. Esta deficiencia de enzimas puede poner en peligro la vida cuando se ven afectados la respiración y los músculos del corazón. La condición es genética, y ambos padres deben llevar el gen mutado para que su hijo lo herede.
Hay dos formas de la enfermedad de Pompe, el inicio infantil y el inicio tardío, que causan debilidad muscular. Cómo progresa la enfermedad depende de cuán temprano comience. Pom Enfermedad de Pompe de inicio infantil
El inicio infantil se considera la forma grave de la enfermedad de Pompe. La condición generalmente aparece dentro de los primeros meses de vida. Los bebés son débiles y tienen problemas para levantar la cabeza. Sus músculos cardíacos se enferman y sus corazones se agrandan y debilitan. También pueden tener lenguas grandes y prominentes y un hígado agrandado.
Otros síntomas incluyen:
Falta de crecimiento y aumento de peso (falta de crecimiento)
Defectos cardíacos y latidos cardíacos irregulares
- Dificultad para respirar, que pueden incluir desmayos
- Problemas para alimentarse y tragar
- Falta de hitos del desarrollo como voltearse o gatear
- Problemas para mover los brazos y piernas
- Pérdida auditiva
- La enfermedad progresa rápidamente, y los niños generalmente mueren por insuficiencia cardíaca y debilidad respiratoria antes de su primer cumpleaños. Los niños afectados pueden vivir más tiempo con intervenciones médicas apropiadas.
- Enfermedad de Pompe de inicio tardío
La enfermedad de Pompe de inicio tardío usualmente comienza con síntomas de debilidad muscular que pueden comenzar en cualquier momento desde la niñez temprana hasta la adultez. La debilidad muscular afecta más a la mitad inferior del cuerpo que a las extremidades superiores. La enfermedad progresa más lentamente que la forma infantil, pero los individuos todavía tienen una expectativa de vida más corta.
La esperanza de vida depende de cuándo comienza la condición y de la rapidez con que progresan los síntomas. Los síntomas como dificultad para caminar o subir escaleras comienzan y progresan lentamente a través de los años. Al igual que con el comienzo temprano, las personas con inicio tardío también pueden desarrollar problemas respiratorios. A medida que la enfermedad avanza, las personas se vuelven dependientes de una silla de ruedas o postradas en la cama y es posible que necesiten un respirador para respirar.
Diagnóstico
La enfermedad de Pompe generalmente se diagnostica después del progreso del síntoma. En los adultos, la enfermedad de Pompe se puede confundir con otras enfermedades musculares crónicas como la esclerosis múltiple. Si su médico sospecha la enfermedad de Pompe, pueden examinar la actividad de la enzima alfa-glucosidasa ácida en las células de la piel cultivadas. En adultos, se puede usar un análisis de sangre para determinar una reducción o ausencia de esta enzima.
Tratamiento
Una persona con la enfermedad de Pompe necesitará atención médica especializada de genetistas, especialistas en metabolismo y neurólogos. Muchas personas encuentran que una dieta alta en proteínas es útil, junto con un amplio ejercicio diario.
Las evaluaciones médicas frecuentes son necesarias a medida que la enfermedad progresa.
En 2006, la Agencia Europea de Medicamentos (EMEA) y la Administración de Alimentos y Medicamentos de los Estados Unidos (FDA) otorgaron la aprobación de comercialización para el medicamento Myozyme para tratar la enfermedad de Pompe. En 2010, Lumizyme fue aprobado. Myozyme es para pacientes menores de 8 años, mientras que Lumizyme está aprobado para personas mayores de 8 años. Ambas drogas reemplazan la enzima faltante, por lo tanto, ayudan a reducir los síntomas de la enfermedad. Tanto Myozyme como Lumizyme se administran por vía intravenosa cada dos semanas.
